PhyML, un logiciel montpelliérain pour retracer l’épidémie du Covid-19
Publié par Université de Montpellier UM, le 11 mai 2020 830
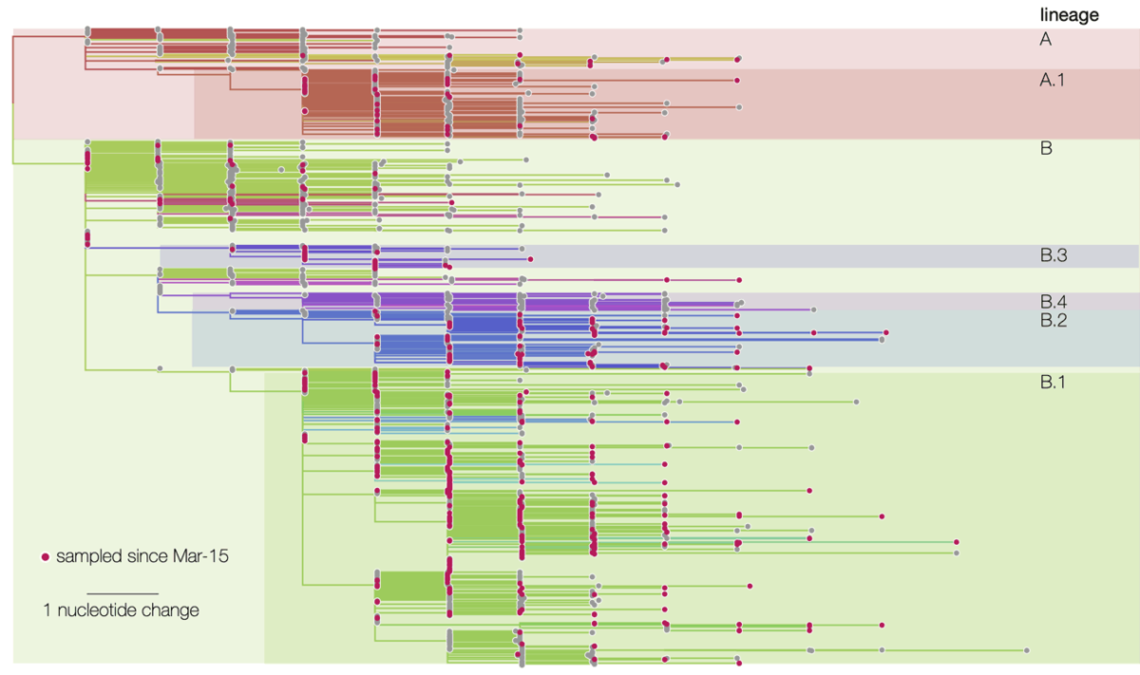
Phylogénie récente du virus SARS-CoV-2
Le logiciel de bio-informatique PhyML, développé depuis une quinzaine d’années au sein du Laboratoire d’informatique, de robotique et de microélectronique de Montpellier (Lirmm) et mis à disposition de la communauté scientifique internationale, est aujourd’hui un outil important pour tracer les origines de la pandémie de Covid-19 et suivre l’évolution du virus.
Avec l’irruption du SARS-CoV-2, virus responsable du Covid-19, le logiciel PhyML, déjà utilisé pour suivre des épidémies virales plus classique telle que la grippe saisonnière, tourne aujourd’hui à plein régime. « Nous travaillons beaucoup, le logiciel évolue en permanence, nous implémentons des algorithmes pour aller plus vite et pour traiter de plus en plus de jeux de données, développer des fonctionnalités nouvelles… » explique Stéphane Guindon, chercheur CNRS au Laboratoire d’informatique, de robotique et de microélectronique de Montpellier (Lirmm).
Développé au cours de sa thèse en 2003 par ce bio-informaticien et son directeur de thèse, Olivier Gascuel, PhyML a été parmi les premiers logiciels gratuits et accessible via la plateforme de bio-informatique hébergée par le Lirmm. « Nous mettons à disposition des laboratoires de recherche nos serveurs car ces analyses phylogénétiques sont très gourmandes en temps de calcul. Certaines peuvent durer des heures, des jours, voire des semaines parfois. » Chaque année la plateforme ATGC réalise ainsi environ 350 000 heures de calcul pour des laboratoires situés en France mais aussi et surtout dans toute l’Europe, en Chine, aux États-Unis…
Retracer une chaîne de transmission
Mais à quoi sert PhyML et pourquoi est-il si utile en temps d’épidémie ? Parce que ce logiciel de bio-informatique est capable de retracer la généalogie, ou phylogénie, de tout ensemble d’organismes pourvu d’un ADN. « Un arbre phylogénétique c’est un peu l’équivalent de l’arbre généalogique qui permet de retracer les liens de parentés d’individus d’une même famille. Cette « généalogie » est reconstruite ici en comparant les génomes de différentes espèces. Dans le cas d’un virus, nous analysons les ~30 000 nucléotides qui constituent son génome afin de retracer une chaîne de transmission. »
Des séquences d’ADN du SARS-CoV-2 ont ainsi été comparées par le logiciel à toutes celles contenues dans des bases de données existantes et ont permis d’établir un lien de proximité fort avec un virus présent chez la chauve-souris et le pangolin. « C’est un virus qui circule chez la chauve-souris mais qui ne provoque pas de mortalité chez cette espèce contrairement à ce qu’on observe chez l’homme à l’heure actuelle » constate Stéphane Guindon. L’analyse de ces séquences a également permis de déterminer que l’épidémie actuelle n’avait eu pour point de départ qu’un seul évènement de zoonose, autrement dit qu’un seul cas de transfert animal-homme. « S’il y avait eu plusieurs évènements l’arbre du virus n’aurait pas la même forme, il présenterait plusieurs sous-arbres, chacun de ces derniers correspondant à un transfert d’une souche virale de l’animal à l’homme. »
Suivre et prévoir l’évolution du virus
La phylogénie à permis de répondre a d’autres questions importantes concernant l’épidémie. « Dès le début du mois de mars, les premières analyses phylogénétiques nous ont indiqué que la taille de population du virus doublait tous les 5 à 7 jours » précise le chercheur. Les analyses phylogénétiques par PhyML pourraient également permettre de mieux prévoir l’évolution du SARS-CoV-2 sur le long cours. Par exemple, chaque année la phylogénie aide à déterminer quelles souches du virus de la grippe sont les plus susceptibles de se développer l’hiver suivant, contribuant ainsi à la conception de vaccins efficaces.


